Inhoud
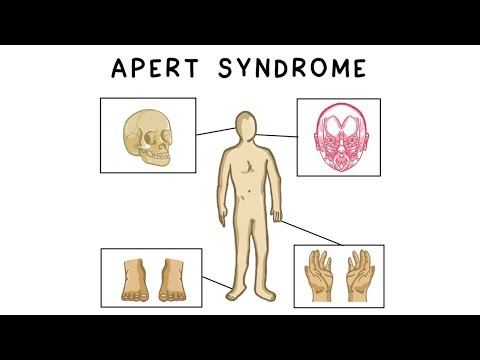
Apert-syndroom is een genetische aandoening die jaarlijks naar schatting één op de 65.000 tot 88.000 pasgeborenen treft. Veel voorkomende kenmerken bij mensen met het Apert-syndroom zijn onder meer voortijdig versmolten schedelbeenderen, versmelting van enkele vingers en tenen. Hoewel deze aandoening een verschillende mate van lichamelijke en verstandelijke beperking veroorzaakt, kunnen mensen met het Apert-syndroom een vol en productief leven gaan leiden.Als u of iemand die u kent een baby met Apert-syndroom verwacht of als u gewoon wat meer over deze aandoening wilt weten, is het altijd nuttig om uzelf uit te rusten met de symptomen, oorzaken, diagnose, behandeling en coping.
Symptomen
Baby's met het Apert-syndroom hebben bepaalde schedelbeenderen die voortijdig samensmelten in utero (vóór de geboorte), waardoor een aandoening ontstaat die bekend staat als craniosynostose. Deze vroege versmelting voorkomt dat de schedel naar behoren groeit en beïnvloedt de vorm van het hoofd en het gezicht. Veel voorkomende gelaatstrekken bij mensen met het Apert-syndroom zijn onder meer:
- Verzonken uiterlijk van het gezicht
- Uitpuilende en / of wijd uit elkaar staande ogen
- Snavelneus
- Onderontwikkelde bovenkaak
- Overvolle tanden en andere gebitsproblemen
Omdat de vroege versmelting van de schedel problemen kan veroorzaken voor de zich ontwikkelende hersenen, kunnen mensen met het Apert-syndroom ook cognitieve beperkingen hebben. Het bereik van ontwikkelingsachterstand en verstandelijke beperking varieert enorm, het kan variëren van normaal tot matig.
Andere kenmerken en aandoeningen die kunnen voorkomen bij mensen met het Apert-syndroom zijn:
- Syndactylie (minimaal drie vingers aan elke hand en voet die met zwemvliezen of gefuseerd kunnen zijn)
- Polydactylie (komt minder vaak voor, maar er kunnen extra cijfers op de handen of voeten staan)
- Gehoorverlies
- Overmatig zweten (hyperhidrose)
- Overmatig vette huid en ernstige acne
- Plekjes ontbrekend haar in de wenkbrauwen
- Gespleten gehemelte
- Terugkerende oorontstekingen
- Gesmolten botten in de nek (halswervels)
- Gaten in de ventriculaire wand van het hart
- Slokdarmblokkade
- Slecht geplaatste anus
- Verstopping van de vagina
- Cryptorchisme (falen van de testikels om af te dalen in de scrotumzak)
- Vergroting van de nieren door blokkering van de urinestroom
Oorzaken
Apert-syndroom wordt veroorzaakt door een mutatie in het FGFR2-gen. Volgens de National Institutes of Health in de Verenigde Staten "produceert dit gen een eiwit dat fibroblastgroeifactorreceptor 2 wordt genoemd. Onder zijn vele functies signaleert dit eiwit dat onrijpe cellen bot worden. cellen tijdens de embryonale ontwikkeling Een mutatie in een specifiek deel van deFGFR2 gen verandert het eiwit en veroorzaakt langdurige signalering, wat de voortijdige versmelting van botten in de schedel, handen en voeten kan bevorderen. "
Hoewel deze aandoening genetisch is, komt het bijna altijd voor bij mensen zonder familiegeschiedenis van het Apert-syndroom, wat betekent dat het wordt veroorzaakt door een nieuwe mutatie.
Mensen met het Apert-syndroom kunnen de genen echter doorgeven aan hun kinderen. Als dit gebeurt, wordt de ziekte overgedragen als een autosomaal dominante aandoening.
Diagnose
Artsen kunnen het Apert-syndroom al voor de geboorte vermoeden vanwege een abnormale ontwikkeling van de schedel.De officiële diagnose wordt gesteld door middel van genetische tests, die worden gedaan door middel van een bloedtest. Dit kan worden uitgevoerd door middel van een vruchtwaterpunctie terwijl de moeder nog zwanger is als het Apert-syndroom wordt vermoed.
Het wordt vaak op echografie geïdentificeerd vanwege de abnormaal ontwikkelende schedelbeenderen. Foetale MRI kan echter veel meer details over de hersenen geven dan echografie. Bevestiging van de diagnose wordt gedaan door middel van bloedonderzoek naar het gen dat de diagnose veroorzaakt.
Behandeling
Er is geen behandeling die het Apert-syndroom kan "genezen", aangezien het een genetische aandoening is. Er zijn echter veel therapieën, operaties en andere interventies die de kwaliteit van leven van een persoon met het Apert-syndroom kunnen verbeteren. De specifieke interventies die nodig zijn, zijn afhankelijk van het individu en hoe deze worden beïnvloed.
Veel voorkomende operaties voor kinderen met het Apert-syndroom zijn onder meer:
- Schedel hervormt
- Frontale orbitale vooruitgang (om de ruimte in het voorhoofd en de oogkassen te vergroten)
- Vorderingen in het midden van het gezicht
- Dubbel gelaatsschot om de bovenkaak te verwijden
- Osteotomie (uitzetting van boven- en onderkaak)
- Neuscorrectie (plastische chirurgie van de neus)
- Genioplastiek (plastische chirurgie van de kin of wangen)
- Ooglid operatie
- Scheiding van vingers en / of tenen
- Hartoperatie voor aangeboren hartafwijkingen
Mensen met het Apert-syndroom hebben mogelijk speciale artsen nodig, vooral tijdens hun kinderjaren, om problemen als een gespleten gehemelte en gehoorproblemen te behandelen. Ze kunnen ook baat hebben bij vroege interventiediensten zoals logopedie, ergotherapie en fysiotherapie als ze tekenen van ontwikkelingsachterstand vertonen.
Sommige mensen met het Apert-syndroom hebben een verstandelijke beperking of vertragingen, maar velen kunnen hun leeftijdsgenoten inhalen.
Omgaan
Het hebben van een kind met speciale behoeften kan voor elke ouder overweldigend zijn. Als uw baby de prenatale diagnose van het Apert-syndroom krijgt, overleg dan met uw arts over wat u kunt verwachten. Erfelijkheidsadvies is absoluut aan te raden omdat een erfelijkheidsadviseur niet alleen de oorzaken van het Apert-syndroom kan uitleggen, maar ook kan adviseren over de kansen op het krijgen van extra kinderen met het Apert-syndroom. Verzamel informatie uit betrouwbare bronnen en praat indien mogelijk met andere ouders. Hoewel de diagnose in het begin misschien overweldigend en beangstigend lijkt, kunt u ontdekken dat deze beter beheersbaar is dan u aanvankelijk had verwacht.
Apert-syndroom is een zeldzame aandoening, maar er zijn meerdere bronnen en steungroepen beschikbaar in de Verenigde Staten en de wereld. Met internet en sociale media is het gemakkelijker dan ooit om verbinding te maken met andere gezinnen en steun te vinden. Zoek ook naar craniofaciale centra bij u in de buurt. Hoe meer bronnen u kunt vinden en aansluiten, hoe comfortabeler u zich zult voelen.
Een woord van Verywell
De diagnose Apert-syndroom kan voor iedereen beangstigend en moeilijk zijn. Het is niet iets waar de meeste mensen van hebben gehoord en het kan aanzienlijke medische complicaties veroorzaken. Er zijn echter veel middelen beschikbaar om gezinnen te helpen, zodat kinderen en volwassenen met het Apert-syndroom kunnen leven en gedijen in de wereld van vandaag.
Wat is het Jackson-Weiss-syndroom?