
Inhoud
Cystic fibrosis (CF) is een erfelijke, levensbedreigende aandoening die de longen en het spijsverteringskanaal beschadigt. Het wordt veroorzaakt door een defect gen dat de productie van verdikt slijm veroorzaakt dat de luchtwegen verstopt en de afscheiding van spijsverteringsenzymen blokkeert.De symptomen zijn progressief en vaak ernstig, en kunnen ademhalingsproblemen, terugkerende longinfecties, slechte groei, mannelijke onvruchtbaarheid en chronische ontsteking van de alvleesklier, lever, nieren en hart omvatten.
CF kan worden gediagnosticeerd met bloedonderzoek, genetische screening en een procedure die bekend staat als een zweetchloridetest.
Hoewel er geen remedie is voor CF, zijn er behandelingen die zowel de lengte als de kwaliteit van iemands leven kunnen verbeteren.
Deze omvatten technieken voor het vrijmaken van luchtwegen, geïnhaleerde antibiotica, slijmverdunners, pancreasenzymen, een calorierijk dieet en nieuwere generatie medicijnen die bekend staan als CFTR-modulatoren. In ernstige gevallen kan een longtransplantatie nodig zijn.
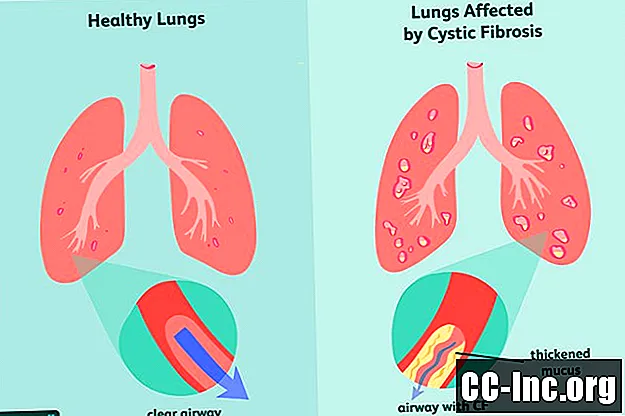
Cystic Fibrosis Symptomen
Als genetische aandoening is cystische fibrose iets waarmee je wordt geboren. Het kan al dan niet symptomen vertonen op het moment van de geboorte en het kan vaak maanden of zelfs jaren duren voordat er tekenen van ziekte verschijnen. Tegen die tijd kunnen de longen en het spijsverteringskanaal al schade hebben opgelopen die niet ongedaan kan worden gemaakt.
De meest voorkomende vroege tekenen en symptomen van CF zijn:
- Blokkering van de eerste ontlasting van de baby (meconium)
- Zout smakende huid
- Een chronische hoest, piepende ademhaling of gekleurd sputum
- Losse, vettige en typisch stinkende ontlasting
- Longinfectie, vaak recidiverend
- Slechte groei en groeiachterstand
Tenzij deze symptomen onder controle kunnen worden gehouden, kan de belasting van de longen (en het onvermogen om aan te komen) een cumulatief effect hebben, waarbij meerdere organen worden aangetast en het risico op complicaties van de ziekte toeneemt.
Enkele van de meer karakteristieke complicaties zijn:
- Vertraagde puberteit
- Bronchiëctasie (de chronische verdikking van de longwanden)
- Gewichtsverlies
- Pancreatitis (ontsteking van de alvleesklier)
- Mannelijke onvruchtbaarheid
- Pulmonale hypertensie (hoge bloeddruk in de longen)
- Galstenen
- Cystic fibrosis-gerelateerde diabetes
- Cor pulmonale (rechtszijdig hartfalen)
- Cirrose (functionele littekens in de lever)
Omdat CF progressieve schade aan cellen en weefsels veroorzaakt, zal elke schade aan de longen en andere organen grotendeels onomkeerbaar zijn. De dood zal meestal het gevolg zijn van ademhalingsfalen, gevolgd door hartfalen en leverfalen.
Symptomen van cystische fibrose
Oorzaken
Cystic fibrosis wordt veroorzaakt door de mutatie van het cystic fibrosis transmembrane receptor (CFTR) -gen, dat verantwoordelijk is voor de productie van het CFTR-eiwit.Dit is het eiwit dat het lichaam nodig heeft om de stroom van zout en water in en uit cellen te reguleren. . Als het eiwit vervormd of defect is, kan het uitdroging op het oppervlak van een cel veroorzaken, wat leidt tot verdikking van het omringende slijm.
CF is een autosomaal recessieve aandoening, wat betekent dat u de CFTR-mutatie van zowel uw moeder als uw vader moet erven om de ziekte te krijgen. Als u slechts één defect gen erft, heeft u geen CF, maar bent u drager van het gemuteerde gen.
U kunt de ziekte erven als elk van uw ouders een CFTR-mutatie heeft of CF zelf. Als beide ouders drager zijn, zou u een:
- 25 procent kans op CF
- 50 procent kans om vervoerder te zijn
- 25 procent kans om niet beïnvloed te worden
Aan de andere kant, als een van je ouders drager is en de ander CF heeft, heb je een kans van 50/50 om CF te hebben of drager te zijn.
Cystic fibrosis is een van de meest voorkomende genetische ziekten en treft ongeveer één op de 2500 baby's die in de Verenigde Staten worden geboren.
Het komt het meest voor bij Kaukasisch en Hispanics, en komt minder vaak voor bij mensen van Afrikaanse of Aziatische afkomst.
Risicofactoren voor cystische fibroseDiagnose
Er zijn een paar tests die worden gebruikt om cystische fibrose te diagnosticeren. Ze werken ofwel door de CFTR-mutatie direct te detecteren of door indirect biologische veranderingen te meten die consistent zijn met de ziekte. De diagnosemethode kan variëren tijdens de zwangerschap, wanneer de baby wordt geboren of op enig moment daarna.
Cystic Fibrosis Doctor Discussiegids
Download onze afdrukbare gids voor uw volgende doktersafspraak om u te helpen de juiste vragen te stellen.

Van de twee standaardtests die vaak worden gebruikt om CF te diagnosticeren:
- Zweet chloride testen, ook gewoon bekend als de zweettest, meet de hoeveelheid chloride op de huid. Omdat CF de overdracht van zout van en naar cellen verstoort, zal er een ophoping van zout in het zweet optreden.
- Genetische CFTR-testen wordt gebruikt om de meest voorkomende mutaties van de CFTR-mutatie te detecteren. Hoewel er meer dan 2000 CFTR-mutaties bekend zijn die cystische fibrose veroorzaken, vertegenwoordigen de 23 in het standaardpanel de meest waarschijnlijke verdachten.
Tijdens de zwangerschap kan de CFTR-genetische test worden gebruikt om vloeistoffen te testen die zijn verkregen via een vruchtwaterpunctie of om cellen te testen die zijn geëxtraheerd via vlokkentest (CVS).
Pasgeboren screening wordt ook standaard gebruikt om CF te diagnosticeren en is tegenwoordig verplicht in alle 50 staten en het District of Columbia. Wat dit inhoudt, hangt af van waar u in de Verenigde Staten woont. Als de resultaten van de screening op pasgeborenen positief zijn, wordt een zweettest gebruikt om de diagnose te bevestigen.
Hoe cystische fibrose wordt vastgesteldBehandeling
Hoewel er geen remedie is voor cystische fibrose, hebben vorderingen in de behandeling de levensduur van degenen die met de ziekte leven verlengd.
Het doel van CF-behandeling is viervoudig: infecties voorkomen, de longfunctie behouden, de spijsvertering normaliseren en de progressie van de ziekte vertragen.
Onder de therapeutische instrumenten die worden gebruikt om CF te behandelen:
- Luchtwegklaringstechnieken (ACT's) worden uitgevoerd om opgehoopt slijm uit de longen te verwijderen en te verdrijven. Technieken zijn onder meer hoesten op de borst, percussie op de borst of oscillatie van de borstwand.
- Een vetrijk, calorierijk dieet wordt gebruikt om de slechte opname van vetten, eiwitten en voedingsstoffen in de darmen te compenseren.
- Pancreasenzymsupplementen worden gebruikt om de spijsverteringsenzymen te ondersteunen die de alvleesklier niet kan produceren vanwege de overmatige ophoping van slijm.
- Antibiotica worden dagelijks ingenomen om bacteriële longinfecties te voorkomen.
- Mucolytica-medicijnen die worden gebruikt om slijm te verdunnen voorafgaand aan ACT's-kunnen worden gebruikt.
- CFTR-modulatoren zijn een nieuwe klasse geneesmiddelen die bepaalde defecten in het CFTR-eiwit kunnen corrigeren en hun regulerende functie kunnen herstellen.
- Zuurstof therapie kan worden gebruikt tijdens acute episodes wanneer uw ademhaling ernstig is gestoord.
- Enterale voeding, ook bekend als sondevoeding, kan worden gebruikt als u niet op gewicht kunt blijven met normale voeding.
- Longtransplantatie wordt overwogen wanneer uw longen niet langer kunnen overleven zonder mechanische ventilatie.
Omgaan
In 1938, toen cystische fibrose voor het eerst als een ziekte werd geclassificeerd, leefden kinderen zelden verder dan hun eerste levensjaar. Tegen de jaren tachtig kon men verwachten 20 tot 25 jaar te leven. Tegenwoordig is het beeld volledig veranderd met mensen die ver in de veertig en zelfs vijftig zijn als de behandeling vroeg wordt gestart en wordt nageleefd.
Dit wil niet zeggen dat CF minder ernstig is dan ooit tevoren. Het is een levensveranderende gebeurtenis, die ijver en consistentie vereist om niet alleen met de ziekte om te gaan, maar ook om de hoogst mogelijke levensstandaard te leven.
Daartoe moet u CF in uw leven normaliseren door de routines en praktijken vast te stellen om de ups en downs te vermijden die stress kunnen veroorzaken en invaliditeit kunnen vergroten. Onder de overwegingen moet u:
- Beheer uw voeding. Mensen met CF hebben vaak tweemaal zoveel calorieën per dag nodig als andere mensen.
- Oefen regelmatig. Fitnessroutines moeten idealiter drie keer per week minimaal 20 tot 30 minuten aërobe activiteit omvatten. Zoek iets leuks dat je een leven lang kunt doen.
- Blijf goed gehydrateerd. Hierdoor blijven de longen en darmen goed werken. Afhankelijk van uw leeftijd moet u niet minder dan zes tot acht grote glazen water per dag drinken.
- Voer een correcte luchtwegklaring uit. Naarmate uw gezondheidsbehoeften veranderen, kunnen ook de soorten opruimingshulpmiddelen die u nodig heeft, veranderen. Praat met uw longarts of fysiotherapeut als u niet de gewenste resultaten behaalt.
- Zoek steun. Naast vrienden en familie kunt u contact opnemen met het dichtstbijzijnde hoofdstuk van de Cystic Fibrosis Foundation (CFF) om verbinding te maken met een ondersteunend netwerk bij u in de buurt.
- Zoek financiële hulp. De CFF biedt diensten die gezinnen helpen om beter om te gaan met de hoge kosten van CF-behandeling.
Een woord van Verywell
Hoewel screenings van pasgeborenen het aantal CF-diagnoses bij baby's dramatisch hebben verhoogd, wordt meer dan 25 procent van de diagnoses alleen gesteld tijdens de kinderjaren, tienerjaren en vroege volwassen jaren.
Dit is problematisch omdat vroege diagnose en behandeling veel van de ernstigere complicaties van CF kunnen voorkomen voordat er ernstige schade kan worden aangericht. Hoewel de behandeling de ziekte niet kan stoppen of ongedaan maken, kan het wel voor veel meer ziektevrije jaren zorgen.
Hiervoor is het belangrijk om de vroege symptomen van CF te kennen en om met uw arts te praten als u vermoedt dat uw kind de ziekte heeft. Dit geldt met name in staten die alleen screenen met IRT-bloedtesten, wat ertoe kan leiden dat maar liefst 5 procent van de kinderen een vertraagde diagnose of een fout-negatief resultaat krijgt, volgens onderzoek van de University of Wisconsin School of Medicine and Public Health .
Welke symptomen kunt u verwachten bij cystische fibrose?